UKCA product marking
New medical device marking requirements and regulations in Great Britain
Since Brexit, additional regulations for medical devices have come into effect in Great Britain, i.e. England, Wales and Scotland. Although the transition periods have been extended, medical device manufacturers should familiarise themselves with the new conformity marking requirements. EU medical device regulations will continue to apply in Northern Ireland.
CE marking serves as part of a product’s declaration of conformity, where manufacturers confirm that all relevant legal requirements have been met. This applies to a range of products, including toys, machinery, electrical appliances and medical devices. Specific directives and regulations apply based on the product category, such as the EU Electromagnetic Compatibility (EMC) Directive for electrical and electronic products.
 A transitional period is in place for surgical instruments, allowing until 30 June 2028 before the UKCA mark becomes mandatory. © freestocks.org I Pexels
A transitional period is in place for surgical instruments, allowing until 30 June 2028 before the UKCA mark becomes mandatory. © freestocks.org I PexelsIn the European Union (EU), the Medical Device Regulation (MDR) applies to medical devices, while the In Vitro Diagnostic Regulation (IVDR) governs in vitro diagnostic medical devices (IVDs). Regardless of the device or IVD class, manufacturers must establish a quality management system in compliance with ISO 13485 as part of the conformity process. Depending on the device or IVD class, manufacturers select the appropriate conformity assessment procedure, which, for all classes except Class I medical devices, requires consultation with a Notified Body. Upon successful completion of the procedure, the manufacturer prepares a declaration of conformity and affixes the CE mark to the product, with the involvement of a Notified Body, if necessary. These procedures remain integral to the regulatory framework even after the transition to the 'new' MDR/IVDR and must be implemented by all medical device manufacturers.
Transition periods have been extended
But what about the sale of medical devices and IVDs in Great Britain (GB), which left the EU in 2020. While the British government announced unrestricted recognition of the CE marking for numerous products, including toys, in August 2023,1) special requirements remain in place for medical devices and IVDs.2)
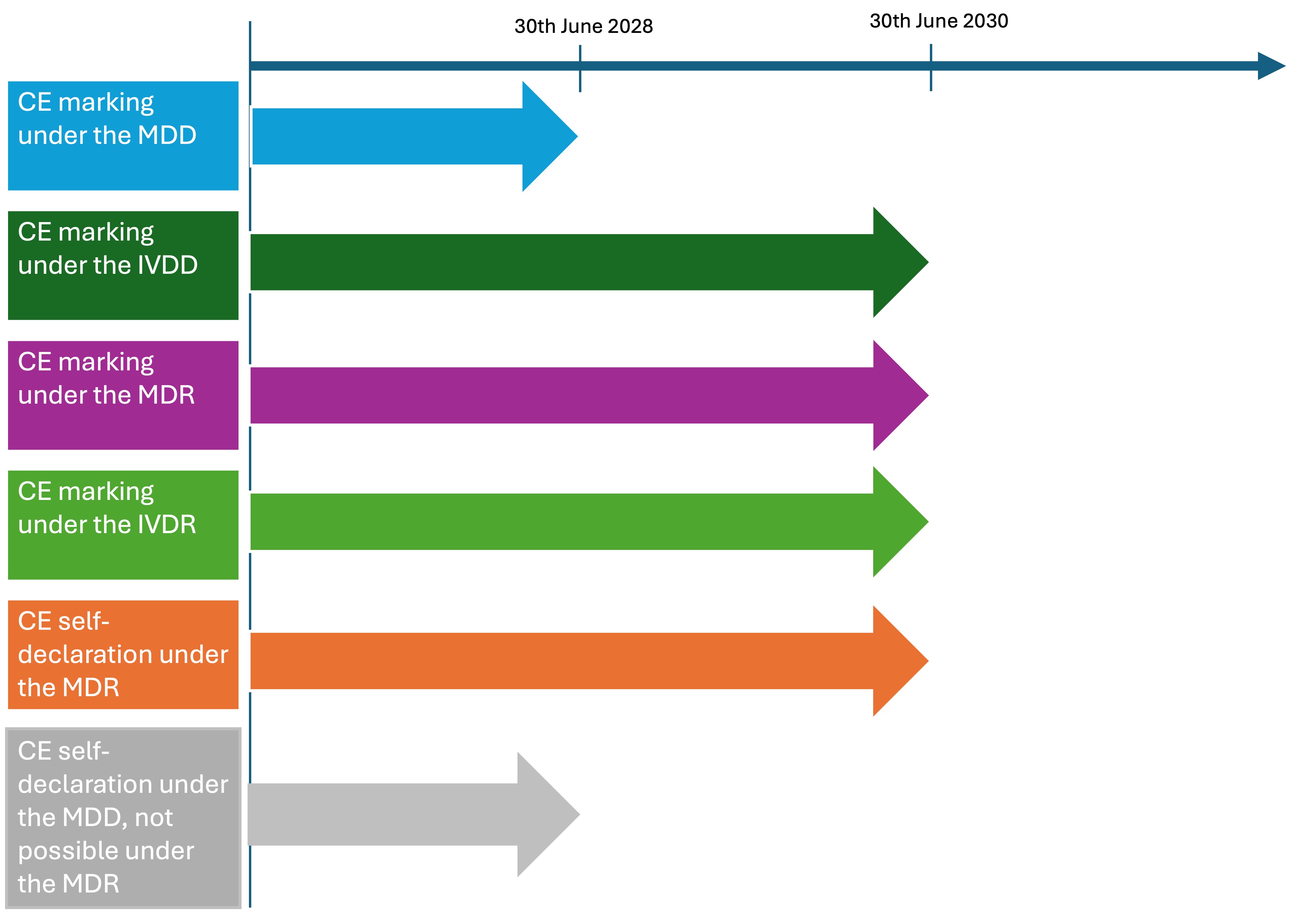
A new regulation for medical devices, the Medical Devices Regulations 2002, is set to come into force in Great Britain in 2025. This regulation makes the United Kingdom Conformity Assessed (UKCA) marking, first introduced in 2021, mandatory. After an extension of the transitional periods, the following deadlines now apply for medical devices with a valid CE marking until the UKCA marking becomes mandatory:3)
- For medical devices that have undergone the conformity procedure under the previous EU Medical Devices Directive (EU MDD), a transitional period is in place until 30 June 2028 or until the relevant certificate expires, whichever is sooner.
- For in vitro diagnostic devices (IVDs) that have followed the conformity procedure under the former EU In Vitro Diagnostics Directive (EU IVDD), a transitional period applies until 30 June 2030 or until the relevant certificate expires, whichever is sooner.
- Medical devices that have undergone the conformity procedure under the new EU Medical Device Regulation (MDR) and in vitro diagnostic devices (IVDs) that comply with the In Vitro Diagnostic Regulation (IVDR) are granted a transitional period until 30 June 2030.
The following rules apply to self-declared CE marked Class I medical devices that were or will be placed on the GB market beyond 30 June 2023:
- Class I medical devices self-declared against EU MDR requirements have a transitional period until 30 June 2030.
- Class I medical devices self-declared against MDD requirements before 26 May 2021 where the involvement of a notified body in their assessment is not required under MDD but is under MDR (such as higher-class devices or reusable surgical instruments), have a transitional period until 30 June 2028. This also applies to Class I devices which have a sterile or measuring function with a valid MDD certificate.
It is important to note that Northern Ireland maintains a special status. For the Northern Irish market, the CE marking remains mandatory if the Notified Body is located within the EU. However, if a British Approved Body was involved in the declaration of conformity of a medical device, the UKNI mark must be affixed alongside the CE mark.4)
 Simplified overview of the transition periods until mandatory use of the UKCA mark
© Dr. Ariane Pott, based on information published on gov.uk
Simplified overview of the transition periods until mandatory use of the UKCA mark
© Dr. Ariane Pott, based on information published on gov.uk
Registration always required
This also means that Class I medical devices and general IVDs, which did not require a Notified Body under the EU MDD or EU IVDD, can only continue to be placed on the GB market during the transition periods if the involvement of a Notified Body is required under the EU MDR or IVDR. This applies to highly classified devices or Class I reusable surgical instruments. Otherwise, UKCA marking must be used for the product. Additionally, customised devices that only comply with the EU MDD or EU AIMDD (old guideline governing active, implantable medical devices) can no longer be placed on the GB market.
Regardless of the type of conformity marking and the applicable transitional periods, manufacturers must register all medical devices and IVDs intended for the GB market the Medicines and Healthcare products Regulatory Agency (MHRA). The registration fee is £240. The registration must be carried out by a UK Responsible Person (UKRP) based in GB, who serves as the primary contact for the MHRA. This individual ensures that the manufacturer follows the conformity procedure, reports serious incidents and supports corrective safety actions. Additionally, the UK Responsible Person is responsible for ensuring that technical documentation and the declaration of conformity are available and accessible when required. Importers must report their activities to the UK Responsible Person, although the ultimate responsibility rests with the manufacturer.5)
Product classes differ
The process for obtaining the UKCA mark depends on the class of medical device.6) Manufacturers of non-sterile and non-measuring Class I devices as well as general IVDs, can self-certify the UKCA mark. For all other device classes, a UK based conformity assessment body must be involved in the conformity assessment process (UK Approved Body). As of September 2024, nine UK approved bodies for medical devices are registered with the MHRA7), including TÜV Süd. 8)
The UKCA conformity procedure distinguishes between the following classes:
- Class I medical devices (based on the classification rules as they apply in GB (Part II of the UK MDR 2002, Annex IX [as modified by Part II of Schedule 2A to the UK MDR 2002])9)
- Class IIa medical devices6)
- Class IIb medical devices6)
- Class III medical devices6)
- In vitro diagnostic medical devices10)
Once the necessary steps for obtaining the UKCA mark are complete, the mark must be clearly visible on the product itself (e.g., an adhesive label), or alternatively, on the packaging or accompanying documentation. Additionally, the contact details of the British importer and the identification number of the Approved Body must be provided.
After the product has been authorised for consumption or use on the GB market, manufacturers are required to conduct market surveillance in collaboration with the UK Responsible Person. This vigilance system involves regular reporting and notification of any adverse events related to the product. Stricter post-market surveillance requirements are anticipated, though specific details are yet to be disclosed.