Produktkennzeichnung UKCA
Neue Kennzeichnungen und Regularien in Großbritannien
Seit dem Brexit treten nun auch weitere Regularien für Medizinprodukte in Großbritannien - England, Wales und Schottland - in Kraft. Auch wenn die Übergangsfristen verlängert wurden, sollten Medizinproduktehersteller sich mit der neuen Konformitätskennzeichnung auseinandersetzen. Für Nordirland gelten weiterhin die CE-Zertifizierungsvorschriften.
Die CE-Kennzeichnung ist Teil der Konformitätserklärung von Produkten. Hersteller erklären damit, dass alle anzuwendenden Rechtsvorschriften für diese Produkte, wie zum Beispiel Spielzeuge, Maschinen, Elektrogeräte und Medizinprodukte, erfüllt werden. Je nach Produkt gelten dabei individuelle Richtlinien und Verordnungen, wie zum Beispiel die EU-Richtlinie für Elektromagnetische Verträglichkeit (EMV) für elektrische oder elektronische Produkte.
 Für chirurgische Instrumente gilt eine Übergangsfrist bis zur verpflichtenden Kennzeichnung mit dem UKCA-Zeichen bis zum 30. Juni 2028. © freestocks.org I Pexels
Für chirurgische Instrumente gilt eine Übergangsfrist bis zur verpflichtenden Kennzeichnung mit dem UKCA-Zeichen bis zum 30. Juni 2028. © freestocks.org I PexelsFür Medizinprodukte gilt dabei in der Europäischen Union (EU) die Medizinprodukteverordnung (MDR) und für In-vitro-Diagnostika (IVD) die Verordnung für In-vitro-Diagnostika (IVDR). Unabhängig von der Klasse des Medizinproduktes bzw. des IVDs muss im Rahmen des Konformitätsverfahrens ein Qualitätsmanagementsystem nach ISO 13485 eingerichtet werden. In Abhängigkeit von der Klasse des Medizinprodukts bzw. IVDs wählt der Hersteller ein bestimmtes Konformitätsbewertungsverfahren aus, welches bei allen Klassen außer der Medizinprodukte Klasse I beinhaltet, dass eine Benannte Stelle hinzugezogen werden muss. Am Ende des Verfahrens erstellt der Hersteller eine Konformitätserklärung und bringt, ggf. unter Einbeziehung der Benannten Stelle, das CE-Zeichen am Produkt an. Diese Verfahren sind auch nach der Umstellung zur „neuen“ MDR/IVDR in der EU etabliert und Teil der Aufgaben der Medizinproduktehersteller.
Übergangsfristen verlängert
Doch wie verhält es sich mit dem Vertrieb von Medizinprodukten und IVDs im ehemaligen EU-Land Großbritannien (GB)? Während die britische Regierung im August 2023 für zahlreiche Produkte wie z. B. Spielzeuge die unbeschränkte Anerkennung der CE-Kennzeichnung bekanntgab,1) gelten für Medizinprodukte und IVDs weiterhin besondere Anforderungen.2)
Ab 2025 soll in GB eine neue Regelung für Medizinprodukte (Medical Devices Regulations 2002) in Kraft treten. Die Regelung beinhaltet auch, dass die bereits im Jahr 2021 eingeführte Konformitätskennzeichnung, die United Kingdom Conformity Assessed-Kennzeichnung (UKCA-Kennzeichnung), verpflichtend wird. Nach einer Verlängerung der Übergangsfristen gelten nun für Medizinprodukte mit einer gültigen CE-Kennzeichnung folgende Fristen bis zu einem verpflichtenden Einsatz der neuen UKCA-Kennzeichnung:3)
- Für Medizinprodukte, die das Konformitätsverfahren unter alter Medizinprodukte-Richtlinie der EU (EU-MDD) durchlaufen haben, gilt eine Übergangsfrist bis zum 30. Juni 2028 oder bis zum Ablauf des Zertifikats - je nachdem, was früher eintritt.
- Für IVDs, die das Konformitätsverfahren unter alter In-Vitro-Diagnostik-Richtlinie der EU (EU-IVDD) durchlaufen haben, gilt eine Übergangsfrist bis zum 30. Juni 2030 oder bis zum Ablauf des Zertifikats - je nachdem, was früher eintritt.
- Für Medizinprodukte, die das Konformitätsverfahren unter der neuen MDR der EU durchlaufen haben, und IVDs, die den Regeln der IVDR entsprechen, gilt eine Übergangsfrist bis zum 30. Juni 2030.
Für Medizinprodukte der Klasse I mit selbstdeklarierter CE-Kennzeichnung, die nach dem 30. Juni 2023 in GB in Verkehr gebracht wurden bzw. werden, gelten folgende Regeln:
- Für Medizinprodukte der Klasse I mit einer Selbstdeklaration gemäß den MDR-Anforderungen der EU gilt eine Übergangsfrist bis zum 30. Juni 2030.
- Für Medizinprodukte der Klasse I, die vor dem 26. Mai 2021 gemäß den MDD-Anforderungen ohne eine Benannte Stelle gekennzeichnet wurden, und die gemäß der MDR die Beteiligung einer Benannten Stelle bei der CE-Kennzeichnung erfordern, wie zum Beispiel höher eingestufte Produkte und wiederverwendbare chirurgische Instrumente, gilt eine Übergangsfrist bis zum 30. Juni 2028. Gleiches gilt für Medizinprodukte der Klasse I mit einem gültigen MDD-Zertifikat, die eine sterile oder messende Funktion haben.
Zu beachten ist, dass Nordirland weiterhin einen Sonderstatus innehat. Für den nordirischen Markt bleibt die Verwendung der CE-Kennzeichnung verpflichtend, wenn die Benannte Stelle in der EU liegt. Wurde ein britischer „Approved Body“ bei der Konformitätserklärung hinzugezogen, muss zusätzlich zur CE-Kennzeichnung die UKNI-Kennzeichnung hinzugefügt werden.4)
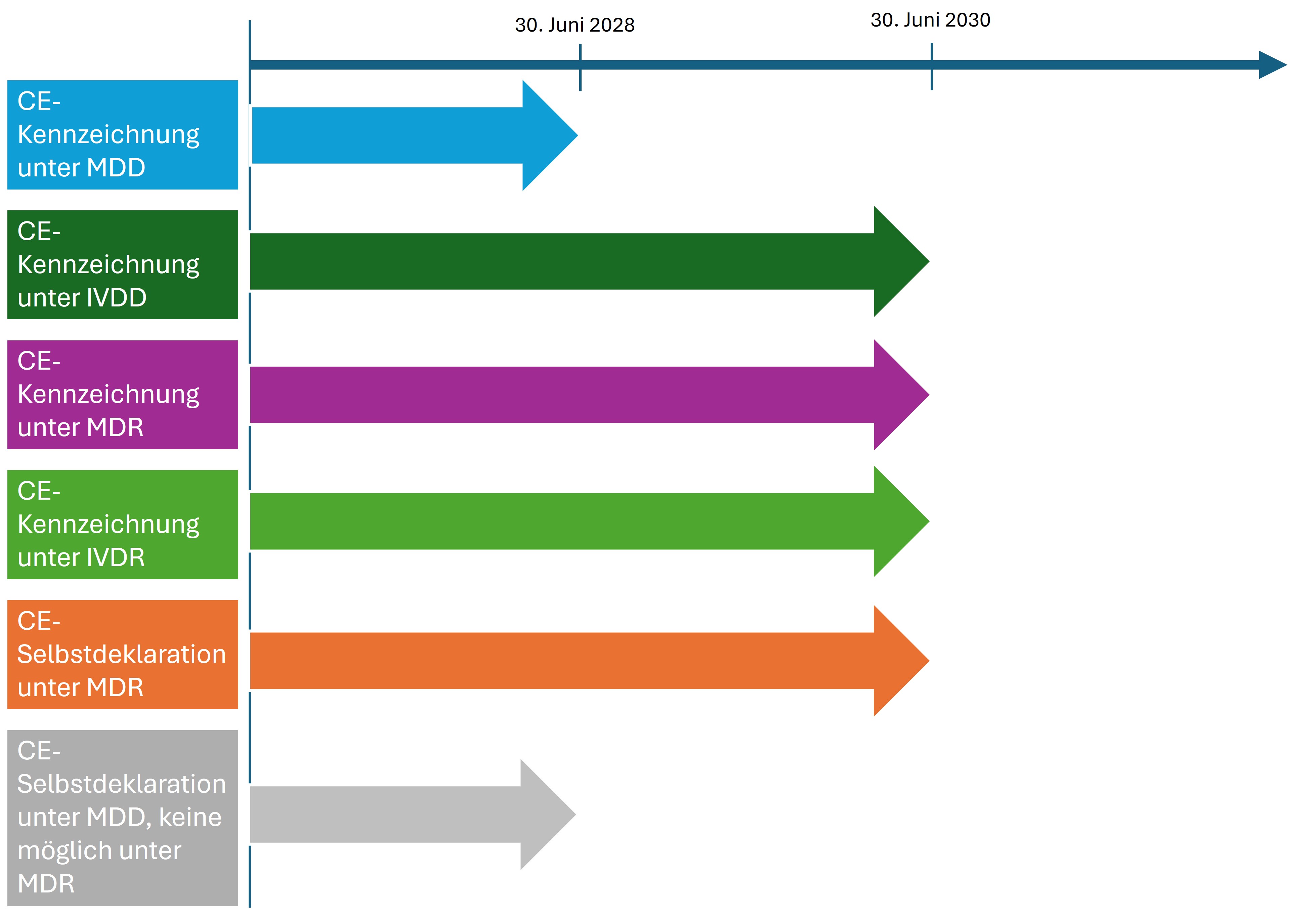 Vereinfachte Übersicht zu den Übergangsfristen bis zur verpflichtenden Nutzung des UKCA-Zeichens
© Dr. Ariane Pott nach Information von gov.uk
Vereinfachte Übersicht zu den Übergangsfristen bis zur verpflichtenden Nutzung des UKCA-Zeichens
© Dr. Ariane Pott nach Information von gov.uk
Registrierung immer erforderlich
Das bedeutet aber auch, dass Medizinprodukte der Klasse I und allgemeine IVDs, für die die Konformitätsbewertung nach der EU-MDD oder EU-IVDD keine Benannte Stelle erforderte, nur dann im Rahmen der Übergangsfristen weiterhin auf dem britischen Markt in Verkehr gebracht werden dürfen, wenn die Einbeziehung einer Benannten Stelle nach der EU-MDR oder IVDR erforderlich wäre. Es muss sich also um ein hochklassifiziertes Gerät oder ein wiederverwendbares chirurgisches Instrument der Klasse I handeln. Ansonsten muss für das Produkt die UKCA-Kennzeichnung verwendet werden. Zudem dürfen Sonderanfertigungen, die nur den Anforderungen der EU-MDD oder EU-AIMDD (alte Richtlinie über aktive implantierbare medizinische Geräte) entsprechen, nicht mehr auf dem Markt in GB in Verkehr gebracht werden.
Unabhängig von der Art der Konformitätskennzeichnung und den Übergangsfristen müssen Hersteller alle Medizinprodukte und IVDs, die in GB in Verkehr gebracht werden sollen, bei der Medicines and Healthcare products Regulatory Agency (MHRA) registrieren. Es fallen Kosten von 240 Britischen Pfund an. Die Registrierung muss durch eine in GB ansässige UK Responsible Person durchgeführt werden. Die Person ist der primäre Ansprechpartner für die MHRA, gewährleistet u. a. die Durchführung des Konformitätsverfahrens durch den Hersteller und meldet schwerwiegende Vorfälle. Sicherheitskorrekturmaßnahmen werden durch die UK Responsible Person unterstützt. Ferner stellt sie sicher, dass eine technische Dokumentation sowie die Konformitätserklärung vorliegen und ist verpflichtet, diese bei Bedarf zur Verfügung zu stellen. Importierende berichten über ihre Tätigkeit an sie. Verantwortlich bleibt jedoch weiterhin der Hersteller.5)
Produktklassen unterscheiden sich
Wie Hersteller die UKCA-Kennzeichnung erhalten, hängt von der Klasse des Medizinproduktes ab.6) Hersteller von nichtsterilen und nichtmessenden Klasse-I-Geräten und allgemeinen IVDs können die UKCA-Kennzeichnung selbst zertifizieren. Für alle weiteren Klassen muss eine in UK zugelassene Stelle eingeschaltet werden (UK Approved Body). Stand September 2024 sind neun Approved Bodies für Medizinprodukte beim MHRA hinterlegt,7) u. a. der TÜV Süd.8)
Für das UKCA-Konformitätsverfahren wird nach folgenden Klassen unterschieden:
- Medizinprodukte der Klasse I (basierend auf der wie in Teil II der britischen MDR 2002, Anhang IX (in der durch Teil II des Anhangs 2A der britischen MDR 2002) geänderten Fassung)9)
- Medizinprodukte der Klasse IIa6)
- Medizinprodukte der Klasse IIb6)
- Medizinprodukt der Klasse III6)
- Medizinische In-vitro-Diagnostika10)
Sind die notwendigen Schritte durchlaufen worden, muss die UKCA-Kennzeichnung gut lesbar auf dem Produkt selbst, z. B. mit einem Klebeetikett, oder auf der Verpackung bzw. den Begleitdokumenten angebracht werden. Zudem müssen die Kontaktdaten des britischen Importierenden sowie die Nummer des Approved Bodies angegeben werden.
Ist das Produkt einmal auf dem britischen Markt zugelassen, sind die Hersteller gemeinsam mit der UK Responsible Person zur Marktüberwachung verpflichtet. Teil dieses Vigilanz-Systems sind regelmäßige Reports sowie Meldungen zu unerwünschten Vorkommnissen mit dem Produkt. Es sind verschärfte Anforderungen an die Überwachung nach dem Inverkehrbringen geplant, Details sind jedoch noch nicht bekannt.